1.1 仪器
本实验使用 LCMS-8045 三重四极杆液质联用系统。具体配置为 LC-30AD 输液泵,DGU-20A5 在线脱气机,SIL-30AC 自动进样器,CTO-20AC 柱温箱,CBM-20A 系统控制器,LCMS-8045 三重四极杆质谱仪,LabSolutions Ver. 5.97 色谱工作站。
1.2 分析条件
液相条件
色 谱 柱:
Shim-pack Velox C18 2.1 mm I.D.× 100 mm L., 2.7 μm
流 动 相:A 相 - 水;B 相 - 乙腈
流 速:0.45 mL/min
柱 温:40℃
进样体积:5 µL
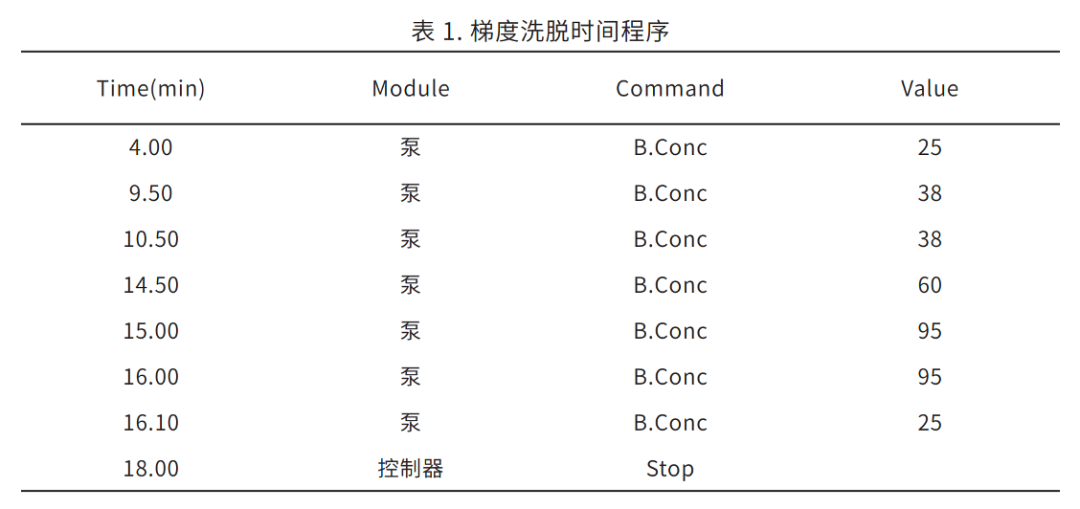
洗脱方式:梯度洗脱,B 相初始浓度为 25%,时间程序见表 1。
质谱条件
离子化模式:ESI(+)
雾化气流速:3.0 L/min
加热气流速:10.0 L/min
干燥气流速:10.0 L/min
接口温度:300℃
加热模块温度:400℃
DL 温度:250℃
碰撞气:氩气
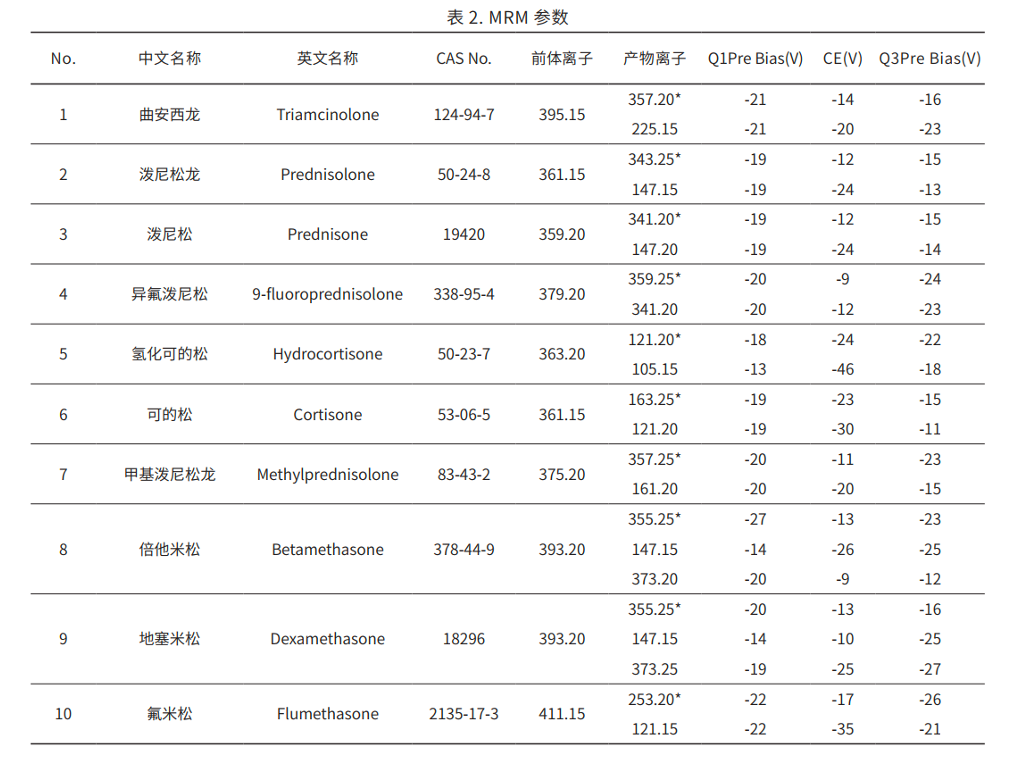
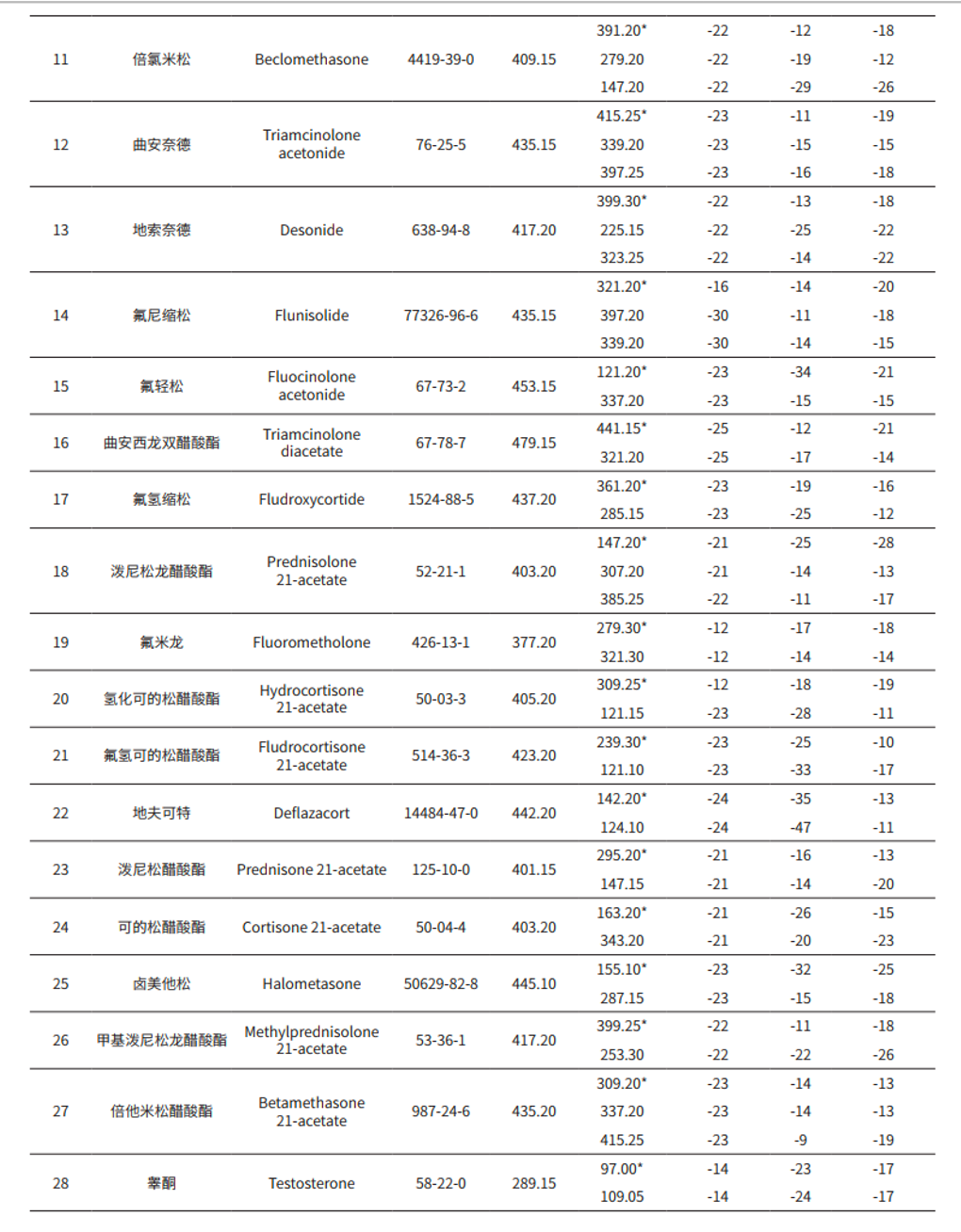
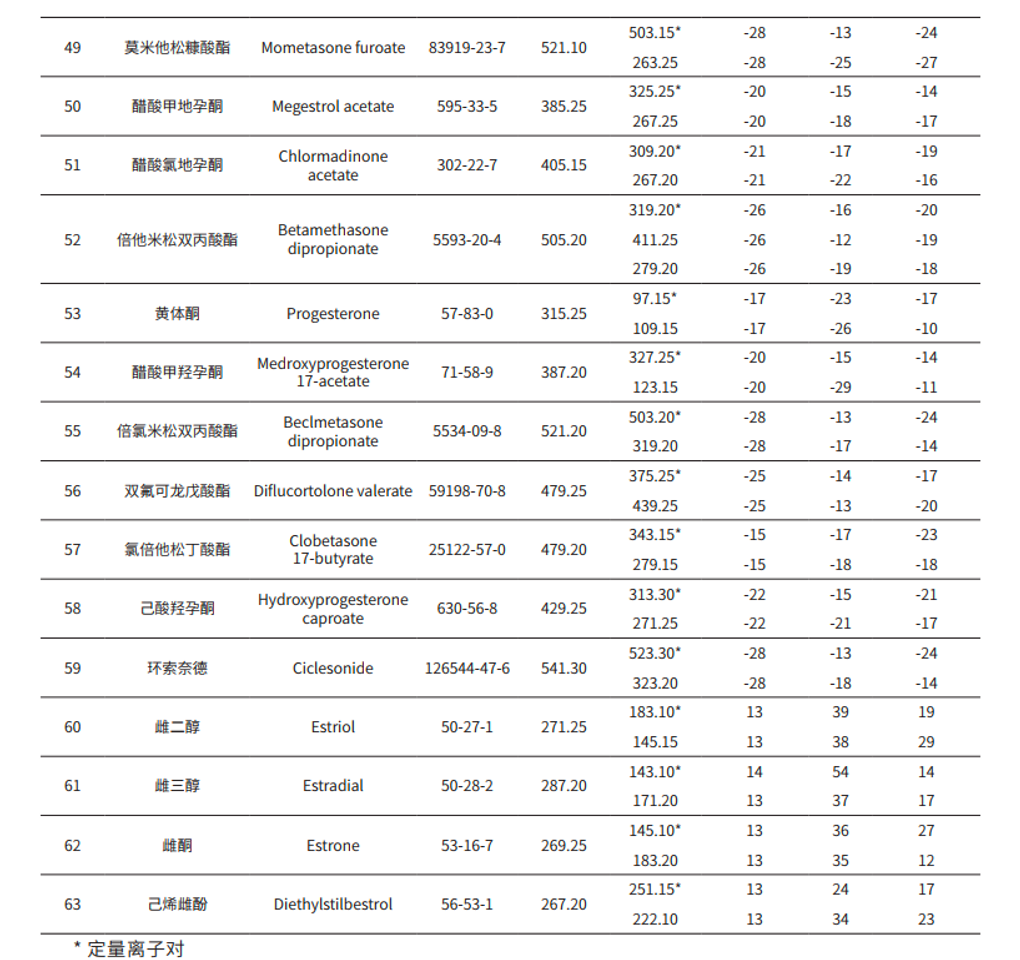
MRM 参数:见表 2
2.1 样品制备
参照“国家药监局 2019 年 第 66 号文件”附件 1 中的激素分析方法。
称取样品 0.2 g(精确到 0.001 g),置于 10 mL 具塞塑料离心管中,加入 2 mL 乙腈,在涡旋混合仪上振荡 30s 至试样与提取溶剂混合均匀,然后再加入 8 mL 乙腈,超声提取 20 min,静置至室温,以 6000 r/min离心 10 min,去取上清液 3 mL 置于 10 mL 离心管,再用 6 mL 水稀释,然后经 0.22 μm 滤膜过滤至 1.5mL样品瓶,备用。
2.2 基质匹配曲线溶液的制备
选取空白乳液样本,经过 2.1 样品前处理制备出空白样品溶液,以此空白样本溶液做溶剂,配制不同浓度的校准曲线溶液,浓度分别为 0.1、0.2、0.5、1.0、2.0 、5.0、10.0、20.0、60.0 和 100.0 μg/L,用于建立基质匹配曲线。
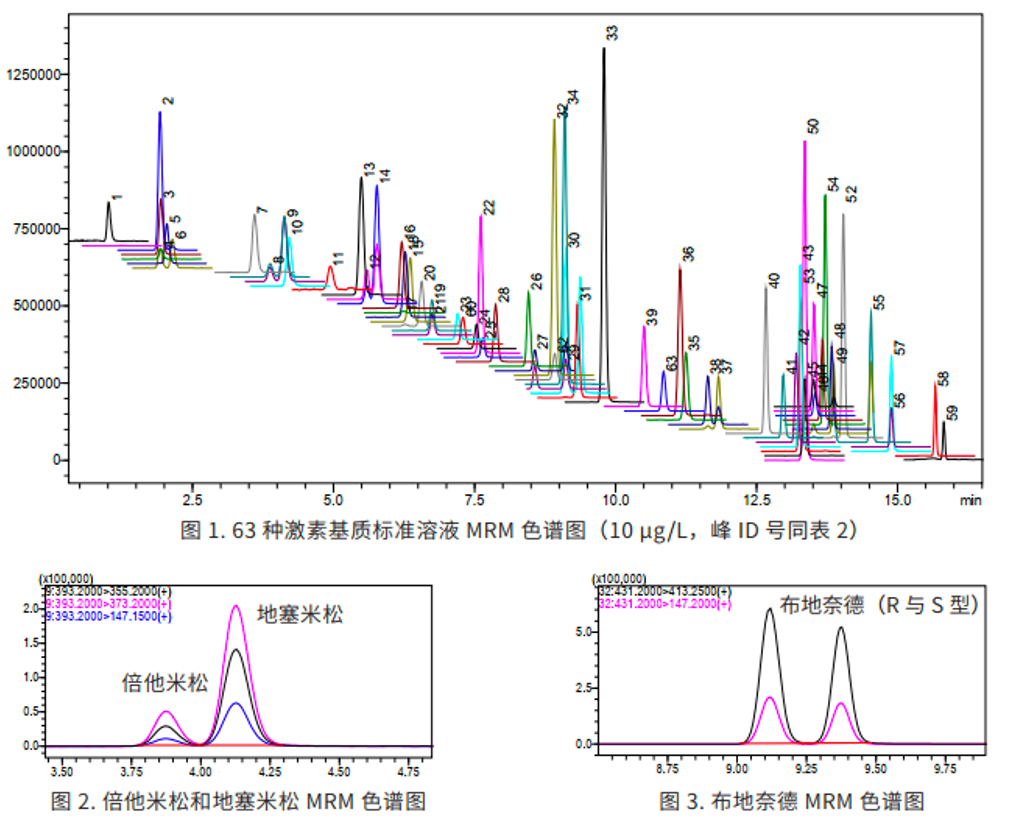
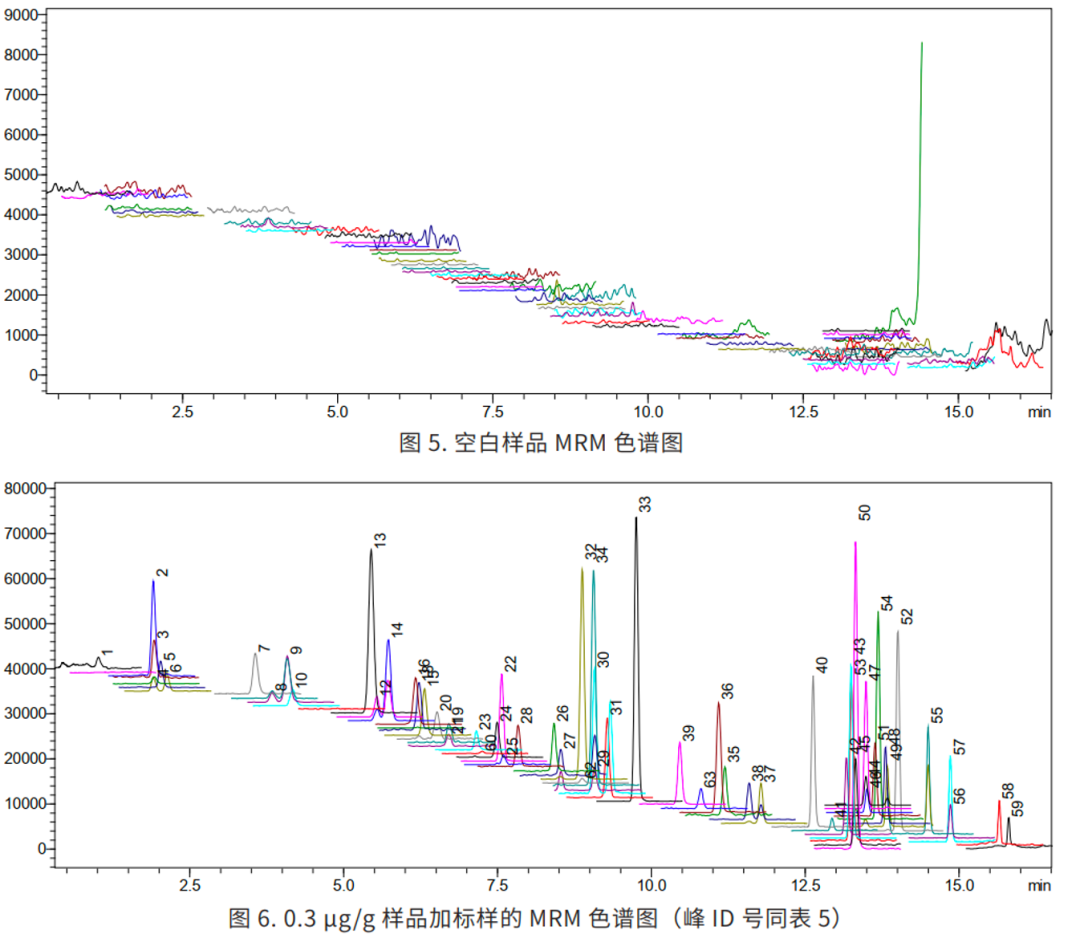
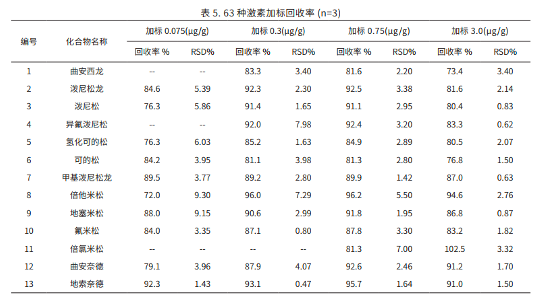
3.1 MRM 色谱图与同分异构体的分离
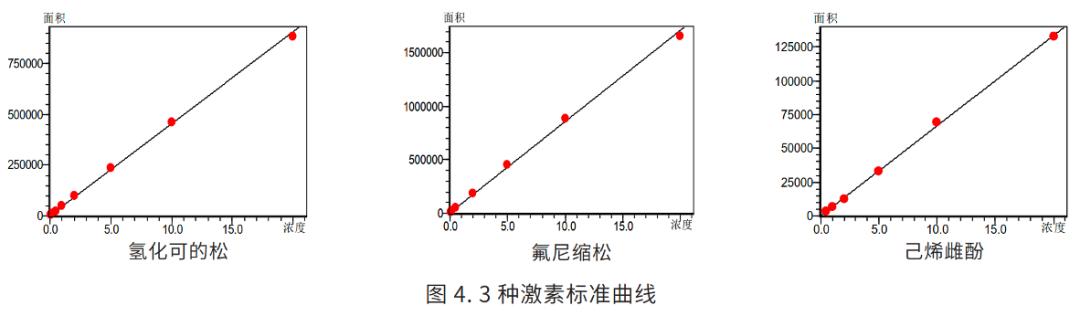
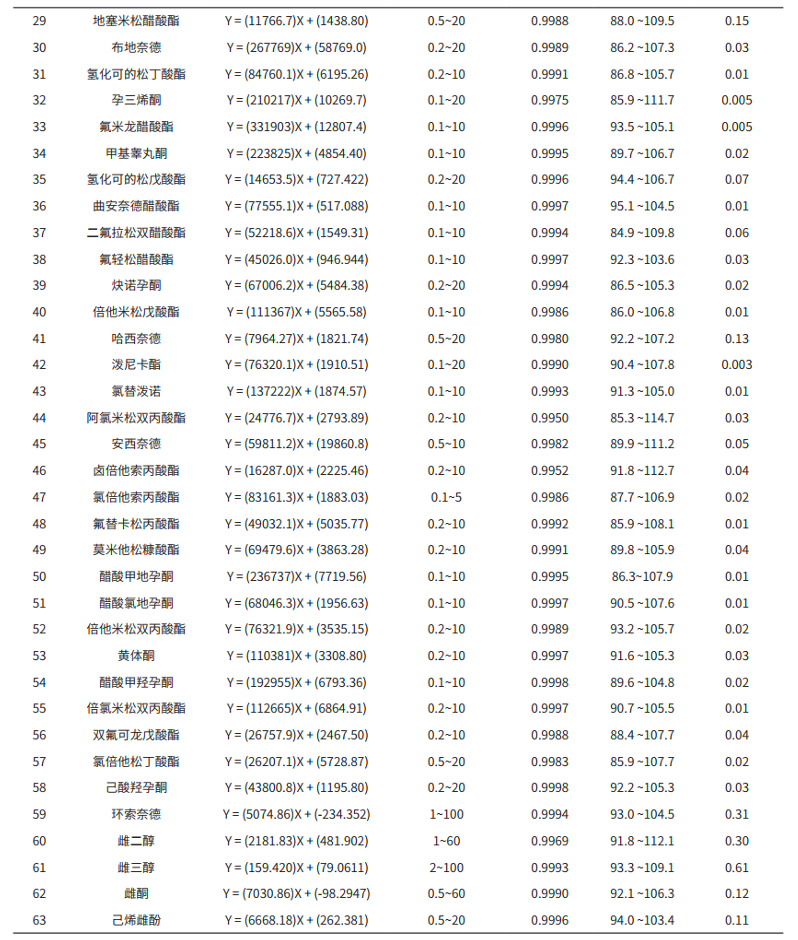
在 63 种激素(MRM 色谱图见图 1)中,有几组物质互为同分异构体,分离难度较大,特别是互为顺反异构的地塞米松和倍他米松(结构见图 2),以及存在 R 型和 S 型的两种差向异构体的布地奈德(Budesonide,简称 BUD,结构见图 4)。本方法选择高柱效的 Velox 色谱柱,将地塞米松和倍他米松(图 2)、布地奈德 R型和 S 型(见图 3)两种同分异构体实现液相分离。
3.2 标准曲线
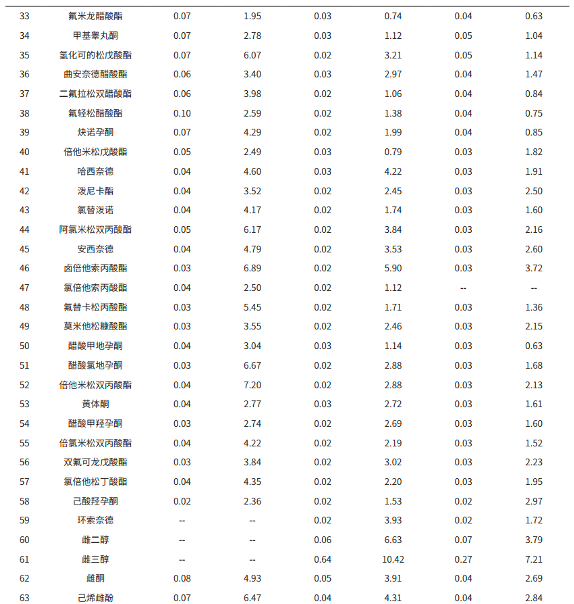
按 1.2 分析条件测定,以浓度为横坐标,峰面积为纵坐标,采用外标法建立基质匹配校准曲线(部分化合物的校准曲线见图 4),63 种激素在相应线性浓度范围内,相关系数在 0.9952~0.9999 之间,各浓度点的回读准确度在 85.0%~112.7% 之间,线性相关性良好。63 种化合物的检出限(ASTM,S/N=3)在 0.002-0.62μg/L 间,满足标准 0.6 μg/L(部分化合物为 2 μg/L)要求,其线性方程、相关系数及仪器检出限见表 3。
3.3 精密度
用空白基质配制不同浓度的混合标准溶液依次进样,每个浓度平行测定 6 次,考察仪器的精密度。结果显示,63 种激素在各浓度下的保留时间和峰面积的相对标准偏差分别在 0.02%~0.64% 和 0.40%~11.85% 之间,结果表明仪器测试的精密度高 ( 见表 4)。
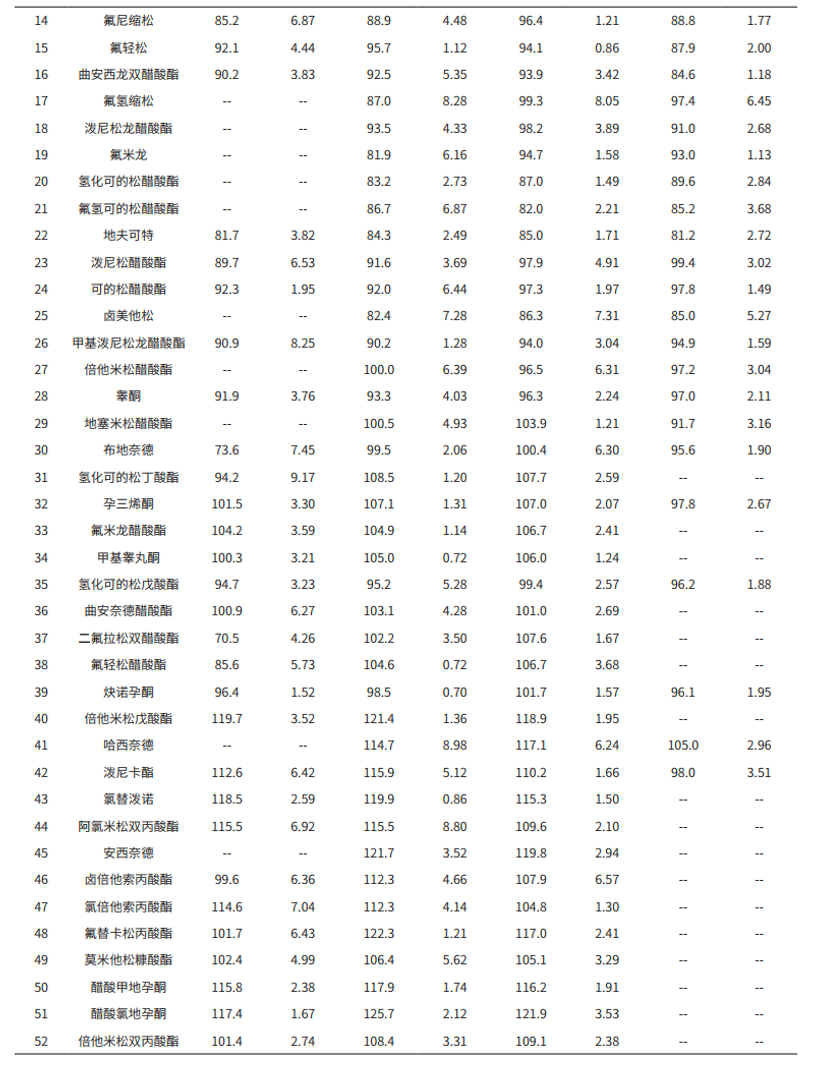
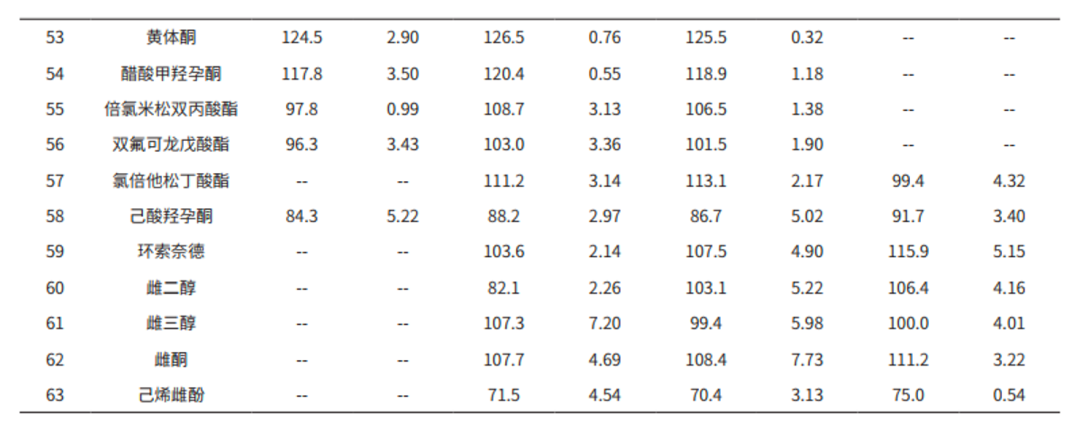
3.4 基质加标实验
称取多份乳液样品各 0.2 g,分别添加 63 种激素标准溶液,参考 2.1 前处理,制备出浓度为 0.075 μg/g、0. 3μg/g、0. 75 μg/g 和 3.0 μg/g 的加标样品。每个浓度平行 3 个,计算 63 种激素在各浓度下的平均加标回收率(见表 5)。结果显示,不同基质加标浓度的回收率范围在 70.4%-126.5% 之间,相对标准偏差在 0.54%-8.98%。
本文使用岛津三重四极杆液质联用仪 LCMS-8045 建立测定化妆品中 63 种激素的分析方法。该方法中 63种激素的基质匹配曲线,其相关系数在 0.9952~0.9999 之间,各浓度点的回读准确度在 85.0%~112.7% 之间,线性相关性良好。63 种化合物的检出限(ASTM,S/N=3)在 0.002-0.62 μg/L 间,满足标准 0.6 μg/L(部分化合物为 2 μg/L)要求。稳定性考察中,63 种激素的保留时间相和峰面积的相对标准偏差分别在 0.02%~0.64%和 0.40%~11.85% 之间,仪器精密度良好。不同基质加标浓度的回收率范围在 70.4%-126.5% 之间,相对标准偏差在 0.54 %-8.98%,符合化妆品实际测试情况。该方法分析速度快,灵敏度高,可为化妆品质量监测等相关行业的从业人员参考使用。