
使用溶剂聚焦功能需要注意的问题 — 相容性

概 述
使用气相色谱仪测定样品时,通过优化色谱分析条件,使得组分出峰宽度变窄的方法被称为“聚焦”,常用的方法有热聚焦、溶剂聚焦和固定相聚焦等。使用不分流进样方式测定低浓度样品时,溶剂聚焦是较为常用的手段。
但分析工作者需要注意样品溶剂与色谱柱固定相之间的“相容性”问题,两者如果不能相容,那么溶剂聚焦的效果反而会变差,此种情况下建议修改分析条件、维护系统或者更换溶剂。
色谱分析案例
我们先以典型的气相色谱分析案例,对溶剂聚焦的问题予以说明。
某第三方环境检测公司使用GCMS-TQ8050型气相色谱-质谱联用仪开展多种农药残留检测工作,其色谱-质谱分析条件如表1所示。

色谱柱箱程序升温曲线如图1所示:
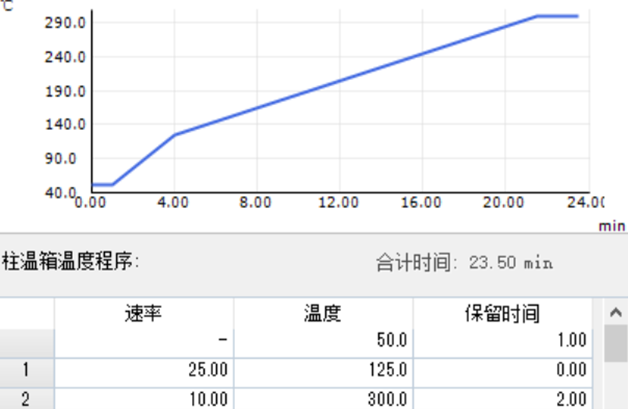
图1:程序升温曲线
用户采用此方法测定以甲醇为溶剂的农残标准样品时,发现保留时间较短的组分出现了峰形分叉的现象(保留时间低于14min的组分),如图2所示。
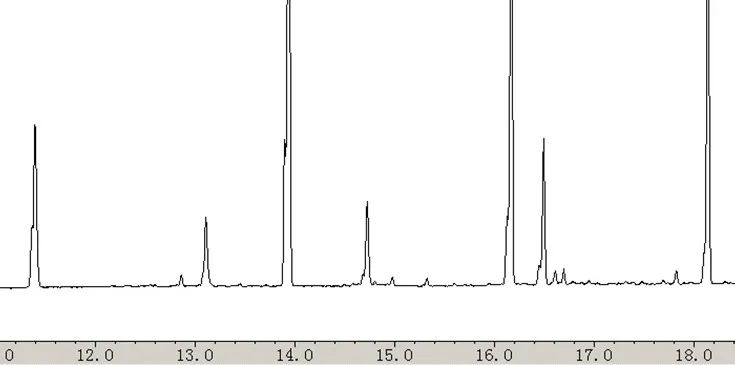
图2:不良峰形的谱图
将此样品的溶剂改换为正己烷之后再次进样,弱保留组分的色谱峰形状则变得尖锐、无分叉——此现象的根本原因与分析条件使用了溶剂聚焦功能有关。
该分析条件的重要特点是柱箱初始温度较低,这就是分析方法使用溶剂聚焦的主要特征——降低色谱柱的初始温度,一般低于样品溶剂沸点10度以下。
溶剂聚焦原理
图3为分流/不分流进样口工作于不分流进样状态下的流量分布示意图。在不分流进样方式下,样品于衬管内气化之后向色谱柱运行的速度比较低(示例图中为1mL/min),从而造成样品所有组分的“起始谱带”较宽,那么就可能导致组分色谱峰出现宽度较大、拖尾严重等不良现象。不良的色谱峰形会导致较差的定量检出限和分辨率,所以通常情况下需要使用辅助手段用以降低峰宽,即“聚焦”功能。
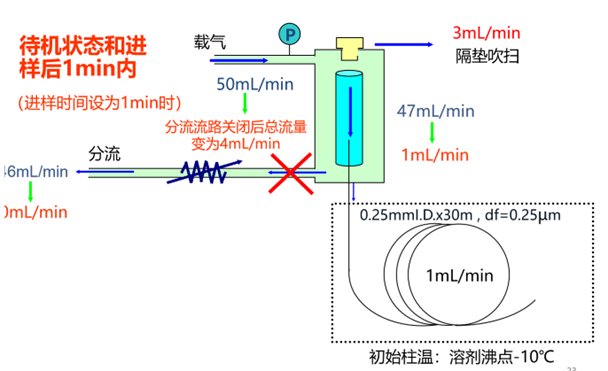
图3:不分流进样方式下进样口的流量分布
溶剂聚焦是较为常用的手段,实现溶剂聚焦的方法比较简单——将色谱柱温度降低到样品溶剂沸点10℃以下。
图4为使用和不使用溶剂聚焦功能对色谱峰形的影响比较结果。左图为不使用溶剂聚焦功能(色谱柱箱初始温度显著高于样品溶剂沸点)谱图,其中保留时间6-8min的组分的色谱峰宽度较大并出现拖尾和分叉问题,右图中对应组分的色谱峰形状良好,同时获得更好的检出限和理论塔板数,显然右图对于色谱分析定量是比较有利的。
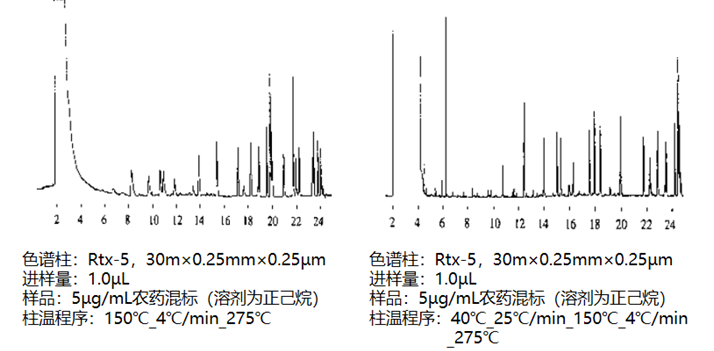
图4:溶剂聚焦对峰形的影响
样品在进样口缓慢气化后进入色谱柱,由于色谱柱初始温度显著低于样品溶剂沸点,样品会在色谱柱内产生冷凝,在色谱柱固定相内表面出现一定长度的液体分布区域(图5中的区域A)。在载气的推动下,沸点较低的溶剂和挥发性组分会慢慢气化和向前运行,沸点较高的目标组分仍然溶解在溶剂中。
经过一段时间的运行,液态样品的空间分布区域就会变得比较窄(图5中的区域B),此时将柱温迅速升高,样品在较窄区域、较短的时间内迅速气化,最终在检测器端就会获得峰宽较窄的色谱峰,整个过程被称为溶剂聚焦。
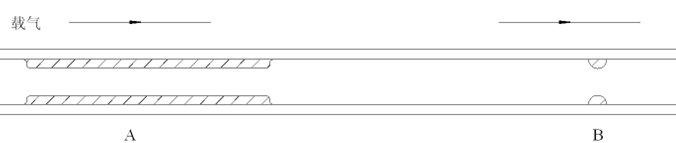
图5:溶剂聚焦图解
溶剂和固定相的相容问题
溶剂聚焦功能的使用似乎比较容易,但是在实际实验中,需要特别注意溶剂选择的问题——应当使用与色谱柱固定相“相容”的样品溶剂。
图1案例中不良色谱图产生的原因是溶剂与色谱柱固定相(极性较强的甲醇和极性较弱的Rtx-5色谱柱)不相容,换言之,溶剂不能良好地浸润固定相表面。如图6所示,如果溶剂不能良好的浸润固定相,那么样品将难以在色谱柱内部形成连续并且厚度均匀的液膜,而倾向于在色谱柱内呈现断续状态分布(图6中区域A所示)。与图5相比,样品运行相同时间后区域B的分布也依然可能呈现断续状态分布,最终使得保留时间较弱组分的色谱峰形出现分叉现象(如图1中的色谱峰状态)。

图6:溶剂不良时色谱柱内样品的分布状态
小结与讨论
本案例中用户更换非极性的正己烷做溶剂进行再次实验,正己烷与Rtx-5固定相的相容性较好,两者可以实现良好聚焦,最终使得色谱峰形状得到改善。
除去更换溶剂的方法以外,还可以考虑修改程序升温,适度延长程序升温的低温时间,也可以减轻或消除色谱峰分叉现象。此外,如果色谱柱由于污染或者固定相损坏等问题,造成固定相(尤其是色谱柱连接进样口部分)化学性质发生变化,也会发生溶剂与固定相不相容问题,最终导致色谱峰形变差。
若实际工作环境无法满足更换溶剂的要求,建议适度提高色谱柱初始温度(略高于溶剂沸点),改用固定相聚焦的方法,以避免不分流进样方式导致的色谱峰形变差。


